This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Study explains why certain immunotherapies don't always work as predicted
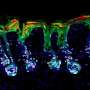
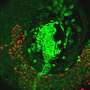
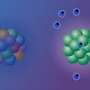
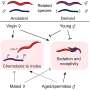
 have accumulated primarily in the supportive tissues (pink regions), while very few have infiltrated tumor cells (islands surrounded by the supportive tissues). Credit: Courtesy of the researchers")
This block is broken or missing. You may be missing content or you might need to enable the original module.
Cancer drugs known as checkpoint blockade inhibitors have proven effective for some cancer patients. These drugs work by taking the brakes off the body's T cell response, stimulating those immune cells to destroy tumors.
Some studies have shown that these drugs work better in patients whose tumors have a very large number of mutated proteins, which scientists believe is because those proteins offer plentiful targets for T cells to attack. However, for at least 50 percent of patients whose tumors show a high mutational burden, checkpoint blockade inhibitors don't work at all.
A new study from MIT reveals a possible explanation for why that is. In a study of mice, the researchers found that measuring the diversity of mutations within a tumor generated much more accurate predictions of whether the treatment would succeed than measuring the overall number of mutations.
If validated in clinical trials, this information could help doctors to better determine which patients will benefit from checkpoint blockade inhibitors.
"While very powerful in the right settings, immune checkpoint therapies are not effective for all cancer patients. This work makes clear the role of genetic heterogeneity in cancer in determining the effectiveness of these treatments," says Tyler Jacks, the David H. Koch Professor of Biology and a member of MIT's Koch Institute for Cancer Research.
Jacks; Peter Westcott, a former MIT postdoc in the Jacks lab who is now an assistant professor at Cold Spring Harbor Laboratory; and Isidro Cortes-Ciriano, a research group leader at EMBL's European Bioinformatics Institute (EMBL-EBI), are the senior authors of the paper, which appears today in Nature Genetics.
A diversity of mutations
Across all types of cancer, a small percentage of tumors have what is called a high tumor mutational burden (TMB), meaning they have a very large number of mutations in each cell. A subset of these tumors has defects related to DNA repair, most commonly in a repair system known as DNA mismatch repair.
Because these tumors have so many mutated proteins, they are believed to be good candidates for immunotherapy treatment, as they offer a plethora of potential targets for T cells to attack. Over the past few years, the FDA has approved a checkpoint blockade inhibitor called pembrolizumab, which activates T cells by blocking a protein called PD-1, to treat several types of tumors that have a high TMB.
However, subsequent studies of patients who received this drug found that more than half of them did not respond well or only showed short-lived responses, even though their tumors had a high mutational burden. The MIT team set out to explore why some patients respond better than others, by designing mouse models that closely mimic the progression of tumors with high TMB.
These mouse models carry mutations in genes that drive cancer development in the colon and lung, as well as a mutation that shuts down the DNA mismatch repair system in these tumors as they begin to develop. This causes the tumors to generate many additional mutations. When the researchers treated these mice with checkpoint blockade inhibitors, they were surprised to find that none of them responded well to the treatment.
"We verified that we were very efficiently inactivating the DNA repair pathway, resulting in lots of mutations. The tumors looked just like they look in human cancers, but they were not more infiltrated by T cells, and they were not responding to immunotherapy," Westcott says.
The researchers discovered that this lack of response appears to be the result of a phenomenon known as intratumoral heterogeneity. This means that, while the tumors have many mutations, each cell in the tumor tends to have different mutations than most of the other cells. As a result, each individual cancer mutation is "subclonal," meaning that it is expressed in a minority of cells. (A "clonal" mutation is one that is expressed in all of the cells.)
In further experiments, the researchers explored what happened as they changed the heterogeneity of lung tumors in mice. They found that in tumors with clonal mutations, checkpoint blockade inhibitors were very effective. However, as they increased the heterogeneity by mixing tumor cells with different mutations, they found that the treatment became less effective.
"That shows us that intratumoral heterogeneity is actually confounding the immune response, and you really only get the strong immune checkpoint blockade responses when you have a clonal tumor," Westcott says.
Failure to activate
It appears that this weak T cell response occurs because the T cells simply don't see enough of any particular cancerous protein, or antigen, to become activated, the researchers say. When the researchers implanted mice with tumors that contained subclonal levels of proteins that normally induce a strong immune response, the T cells failed to become powerful enough to attack the tumor.
"You can have these potently immunogenic tumor cells that otherwise should lead to a profound T cell response, but at this low clonal fraction, they completely go stealth, and the immune system fails to recognize them," Westcott says. "There's not enough of the antigen that the T cells recognize, so they're insufficiently primed and don't acquire the ability to kill tumor cells."
To see if these findings might extend to human patients, the researchers analyzed data from two small clinical trials of people who had been treated with checkpoint blockade inhibitors for either colorectal or stomach cancer. After analyzing the sequences of the patients' tumors, they found that patients' whose tumors were more homogeneous responded better to the treatment.
"Our understanding of cancer is improving all the time, and this translates into better patient outcomes," Cortes-Ciriano says. "Survival rates following a cancer diagnosis have significantly improved in the past 20 years, thanks to advanced research and clinical studies. We know that each patient's cancer is different and will require a tailored approach. Personalized medicine must take into account new research that is helping us understand why cancer treatments work for some patients but not all."
The findings also suggest that treating patients with drugs that block the DNA mismatch repair pathway, in hopes of generating more mutations that T cells could target, may not help and could be harmful, the researchers say. One such drug is now in clinical trials.
"If you try to mutate an existing cancer, where you already have many cancer cells at the primary site and others that may have disseminated throughout the body, you're going to create a super heterogeneous collection of cancer genomes. And what we showed is that with this high intratumoral heterogeneity, the T cell response is confused and there is absolutely no response to immune checkpoint therapy," Westcott says.
More information: Peter M. K. Westcott et al, Mismatch repair deficiency is not sufficient to elicit tumor immunogenicity, Nature Genetics (2023). DOI: 10.1038/s41588-023-01499-4
Journal information: Nature Genetics
Provided by Massachusetts Institute of Technology
This story is republished courtesy of MIT News (web.mit.edu/newsoffice/), a popular site that covers news about MIT research, innovation and teaching.