This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Novel method for analyzing protein crystals could open up new avenues for drug discovery
. DOI: 10.1038/s41467-023-36734-3")
A new method for analyzing protein crystals—developed by Cornell University researchers and given a funky two-part name—could open up applications for new drug discovery and other areas of biotechnology and biochemistry.
The development, outlined in a paper published March 3 in Nature Communications, provides researchers with the tools to interpret the once-discarded data from X-ray crystallography experiments—an essential method used to study the structures of proteins. This work, which builds on a study released in 2020, could lead to a better understanding of a protein's movement, structure and overall function.
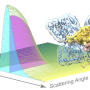
Protein crystallography produces bright spots, known as Bragg peaks, from the crystals, providing high-resolution information about the shape and structure of a protein. This process also captures blurry images—patterns and clouds related to the movement and vibrations of the proteins—hidden in the background of the Bragg peaks.
These background images are typically discarded, with priority given to the bright Bragg peak imagery that is more easily analyzed.
"We know that this pattern is related to the motion of the atoms of the protein, but we haven't been able to use that information," said lead author Steve Meisburger, a former postdoctoral researcher in the lab of Nozomi Ando, associate professor of chemistry and chemical biology in the College of Arts and Sciences. "The information is there, but we didn't know how to use it. Now we do."
Meisburger worked closely with Ando to develop the robust workflow to decode the weak background signals from crystallography experiments called diffuse scattering. This allows researchers to analyze the total scattering from crystals, which depends on both the protein's structure and the subtle blur of its movements.
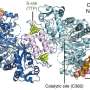
Their two-part method—which the team dubbed GOODVIBES and DISCOBALL—simultaneously provides a high-resolution structure of the protein and information on its correlated atomic movements.
GOODVIBES analyzes the X-ray data by separating the movements—subtle vibrations—of the protein from other proteins that might be moving around it. DISCOBALL independently validates these movements for certain proteins directly from the data, allowing researchers to trust the results from GOODVIBES and understand what the protein might be doing.
Ando said that while the potential for using diffuse scattering has been recognized for a long time, the act of accurately measuring the subtle signal while processing the data for something useful has been very difficult to do.
"It is much more computationally intensive to analyze than trying to analyze crystallography data alone," Ando said. "We have a lot more data to deal with in diffuse scattering, because we are looking everywhere all at once, and the signal is also very nuanced."
The overarching goal, Ando said, is to turn GOODVIBES and DISCOBALL into a genuine structural technique that can be used by researchers at synchrotrons all over the world.
"There is a lot of interest within the structural biology and biochemistry fields to use this signal," Ando said. "We weren't satisfied with just understanding what's contained in the signal; it was really important for us to make the next step of creating the tools, and making GOODVIBES and DISCOBALL available for others to use these tools and test their hypotheses."
These methods were developed using lysozyme proteins collected at the Cornell High Energy Synchrotron Source (CHESS); the Ando group will be returning to CHESS this spring to collaborate with Meisburger, now a CHESS staff scientist, on more complex protein structures using their new method.
By isolating the internal motion signals from total scattering data of these complex proteins, researchers can learn more about how proteins move and interact with other important molecules. This information can be used to design new drugs and therapies that target specific proteins.
More information: Steve P. Meisburger et al, Robust total X-ray scattering workflow to study correlated motion of proteins in crystals, Nature Communications (2023). DOI: 10.1038/s41467-023-36734-3
Journal information: Nature Communications
Provided by Cornell University