This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
A new model offers an explanation for the huge variety of sizes of DNA in nature
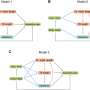
. Three types of solution are possible: exponential growth when pi>pd, linear growth when pi=pd, exponential decay to a steady-state when pi")
A new model developed at Tel Aviv University offers a possible solution to the scientific question of why neutral sequences, sometimes referred to as "junk DNA," are not eliminated from the genome of living creatures in nature and continue to exist within it even millions of years later.
According to the researchers, the explanation is that junk DNA is often located in the vicinity of functional DNA. Deletion events around the borders between junk and functional DNA are likely to damage the functional regions and so evolution rejects them. The model contributes to the understanding of the huge variety of genome sizes observed in nature.
The phenomenon that the new model describes is called by the team of researchers "border induced selection." It was developed under the leadership of the Ph.D. student Gil Loewenthal in the laboratory of Prof. Tal Pupko from the Shmunis School of Biomedicine and Cancer Research, Faculty of Life Sciences and in collaboration with Prof. Itay Mayrose (Faculty of Life Sciences, Tel Aviv University). The study was published in the journal Open Biology.
The researchers explain that throughout evolution, the size of the genome in living creatures in nature changes. For example, some salamander species have a genome ten times larger than the human genome.
Prof. Pupko explains, "The rate of deletions and short insertions, which are termed in short as 'indels', is usually measured by examining pseudogenes. Pseudogenes are genes that have lost their function, and in which there are frequent mutations, including deletions and insertions of DNA segments. In previous studies that characterized the indels, it was found that the rate of deletions is greater than the rate of additions in a variety of creatures including bacteria, insects, and even mammals such as humans. The question we tried to answer is how the genomes are not deleted when the probability of DNA deletion events is significantly greater than DNA addition events."
Ph.D. student Loewenthal says, "We have provided a different view to the dynamics of evolution at the DNA level. As mentioned, when measuring the rate of indels there will be more deletions, but the measurements are carried out in pseudogenes which are quite long sequences. We claim that in shorter neutral segments, deletions are likely to delete adjacent functional segments which are essential for the functioning of the organism, and therefore will be rejected. We call this phenomenon 'border-induced selection'."
"If so, when the segment is short, there will be a reverse bias so that there will be more insertions than deletions, and therefore short neutral segments usually are retained. In our study, we simulated the dynamics of indels, while taking into account the effect of 'border-induced selection' and compared the simulation results to the distribution of human intron lengths (introns are DNA segments in the middle of a protein-coding gene, which themselves do not code for a protein)."
"A good match was obtained between the results of the simulations and the distribution of lengths observed in nature, and we were able to explain peculiar phenomena in the length distribution of introns, such as the large variation in intron lengths, as well as the complex shape of the distribution which does not look like a standard bell curve."
More information: Gil Loewenthal et al, The evolutionary dynamics that retain long neutral genomic sequences in face of indel deletion bias: a model and its application to human introns, Open Biology (2022). DOI: 10.1098/rsob.220223
Journal information: Open Biology
Provided by Tel-Aviv University