Researchers build powerful model for discovering new drugs
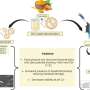
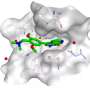
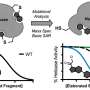
, fingerprints of drugs and targets are calculated from the whole SMILES sequence and the whole amino acid sequence. Global information is extracted from these fingerprints by some fully connected layers. The global information is combined with baseline convolutional neural network models through an attention-like process that guides the convolutional neural network model training. Credit: Big Data Mining and Analytics, Tsinghua University Press")
Researchers have developed a new computer framework that holds promise in the work to discover new drugs. Their framework uses an artificial intelligence method called a convolutional neural network to provide global information about potential novel drug candidates.
The Wuhan University research team published their findings in the journal Big Data Mining and Analytics on November 24, 2022.
The team developed a fingerprint-embedding framework for drug-target binding affinity prediction (FingerDTA), with the capability to find novel drug candidates. The fingerprints (descriptors) of drug and targets are calculated. These targets are molecules that are related in some way to the disease—targets can be useful in the ways drugs are used to fight a particular disease.
Then the team used the general information from the fingerprint of a drug or a target in a convolutional neural network model and promoted its performance in predicting drug-target binding affinity. FingerDTA is a powerful model for discovering new drugs.
Traditional in vivo drug discovery, where researchers work with living subjects to find new drugs for fighting diseases, is a costly, time-consuming process. Researchers can use virtual pre-screening of potential drugs to guide their experiments. This virtual process can reduce costs and improve the success rate in discovering the right drug.
Researchers have widely used two virtual screening methods for drug discovery. One method is high throughput screening, where large compound libraries are tested in a short period of time. Another method involves strategies based on simulated molecular docking, where they study how two or more molecular structures fit together, predicting how a protein interacts with small molecules.
While these two methods have been used successfully in drug discovery, they require in-depth experimental design and verification, making them unsuitable for gigantic-scale drug screening.
A third method uses drug-target affinity prediction models, where scientists look for a strong attraction between the drug and the target as a means of identifying drugs that might be candidates for treating a disease. This third method has great advantages in both efficiency and cost. Scientists have been able to successfully apply deep neural networks to predict the drug-target binding affinity. Therefore, the Wuhan University research team focused their work on a deep learning model for drug-target binding affinity prediction.
Scalability is a major problem when complex algorithms are used to analyze a big data set in terabyte or beyond on a cluster or cloud. The widely used MapReduce type programming model is often used to process large amounts of data across hundreds or thousands of servers. But MapReduce is not scalable to big data because of its memory dependence and high communication costs. The research team has proposed a non-MapReduce computing framework to improve the scalability of cluster computing on big data. Their framework reduces the data communication cost and enables approximate computing, which is less dependent on memory.
"This new computing framework also generates a few benefits in big data computing, such as quickly sampling multiple random samples for ensemble machine learning and approximate computing, directly executing serial algorithms on local random samples without data communications among the nodes, and facilitating big data exploration and cleansing. Besides, non-MapReduce computing simplifies big data computing and can save energy in cloud computing," said Juan Liu, a professor in the School of Computer Science at Wuhan University.
The research team believes that drug-target binding affinity prediction holds promise in discovering new drugs that can inhibit viruses from attaching to their targets. "The FingerDTA can help discover some potential drugs for deactivating COVID-19 by binding to the spike target," said Liu. It can provide precise guidance to save substantial manpower and material resources, while also accelerating new drug research.
Looking ahead, the team hopes to implement the FingerDTA framework in big data platforms and put it in real applications. "Our ultimate goal is to develop such technology and systems for users to tackle the application problems of analyzing extremely big data distributed in several data centers," said Juan Liu.
More information: Xuekai Zhu et al, FingerDTA: A Fingerprint-Embedding Framework for Drug-Target Binding Affinity Prediction, Big Data Mining and Analytics (2022). DOI: 10.26599/BDMA.2022.9020005
Provided by Tsinghua University Press